信頼性の高い遺伝子診断を行うために必要な諸要件
土原:個別化医療のための遺伝子検査は重要ですが、誰でも好きなように検査をしていいわけではありません。保険承認を目指した診断薬開発の道筋はもちろん、その間をつなぐ検査法、あるいは検査基準を策定していくことが大事になってきます。そこで、登先生からゲノム診断における質保証についてお話いただきます。
登:2010年8月に企業側から厚生労働省大臣官房審議官に宛てて「体外診断用医薬品の取り扱いに関する考え方」という文書が出され、そこには、個別化医療においてはコンパニオン診断薬が重要であり、そのための新たな承認審査基準とそのルール作りが喫緊の課題である、と述べられています。米国FDAでは2011年7月にコンパニオン診断薬に関するドラフトガイダンスが出ていますが、日本では2013年7月にようやく厚生労働省から「コンパニオン診断薬等及び関連する医薬品の承認申請に係る留意事項について」という文書が出されました。ここで示されたコンパニオン診断薬の定義は、米国FDAのドラフトガイダンスとほぼ同じ内容です。
その後、2013年12月にPMDAから厚生労働省医薬食品局審査管理課長宛に「コンパニオン診断薬及び関連する医薬品に関する技術的ガイダンス等について」という文書が提出され、厚生労働省から各都道府県宛に事務連絡として通達されました。PMDAが独自の考えでこのようなガイダンスを出すのは異例のことで、コンパニオン診断薬に対する関心の高さがうかがえます。
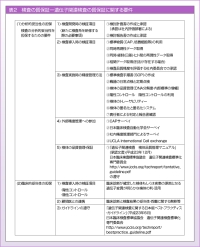
臨床検査を行う側としては、ゲノム診断では、どのように検査の質を保証するかが課題になります。日本衛生検査所協会では、遺伝子検査の質保証体制に関する要件を「検査開発時の検証項目」「検査導入時の検証項目」「検査実施時の精度管理方法」「外部精度管理への参加」「検体の品質管理・保証」の5つに分けて取りまとめています (表2)。「検体の品質管理・保証」は、測定だけでなく検体採取から始まっており、例えば多施設から検体を集積する場合、採取から運搬まで管理する必要があり、厳格に検出したとしても保存状態が悪ければ検査結果に影響してしまいます。
検体の品質管理については、日本臨床検査標準協議会 (JCCLS) の「遺伝子関連検査 検体品質管理マニュアル」が基本になり、同時再現性、日差再現性など必要事項が記載されています。しかし、すべての施設がこれに則った検査を行うのは不可能です。したがって、質管理を徹底したセントラルラボを何ヵ所か準備することが現実的だと思います。
土原:ありがとうございます。臨床検査の質保証は入口から出口まで、すべてがコントロールされていなければならないことを再認識しました。臨床医の立場としては、どこの検査施設に送ればきちんとできるのか、また、自施設で検査したい場合はどうすればいいのかという疑問を持つと思いますが、質保証のステップが細分化されているため、窓口が多いような印象を受けます。検査の認証を受けられるような統一の仕組みはあるのでしょうか。

登:統一された仕組みはありません。臨床医は、すぐに結果を知りたい、検査は手軽に行いたいと思われるでしょうが、コンパニオン診断は治療選択や予後に直結するので慎重に行うべきです。検体の管理から結果の解釈まで含めた精度管理を実現するためには、検査施設をある程度限定したほうがよく、セントラル化することが重要です。
吉野:同感です。検査は片手間でできるものではないことを、臨床医も認識することが大事だと思います。
西尾:セントラルラボは民間の検査施設ですか。
登:現実的には民間になると思います。民間でも厳格な精度管理、人材教育が行われています。
土原:セントラルラボの話が出ましたが、米国にはCLIAラボという施設がありますね。CLIAラボについて教えていただけますでしょうか。
登:かつて米国では、検査施設が乱立し精度管理が問題になったことから、1988年に、臨床検査室改善法 (CLIA: Clinical Laboratory Improvement Amendment) が施行されました。CLIAラボとは、CLIAの認証を受けているラボのことで、自施設内に限り独自に開発した検査法 (LDT: Laboratory Developed Test) を用いて検査を行うことが許されています。
これは、米国において安全性・有効性を管理するFDAと、公的医療保険制度の運営母体であるCMS (Centers for Medicare & Medicaid Services) という2つの組織の狭間で生まれた制度と言えると思います。LDTの使用は自施設内に限られ外に出ないため、実際に精度管理されているかは把握できませんが、CLIAによりラボを認証することで保証している形です。FDAはLDTを規制したいと考えているようですが、一方で、承認されたIVDだけでは進歩の速い臨床のニーズに応えられないため、LDTにより補完されているという側面もあります。
土原:現在は臨床開発のスピードが速いため、承認を受けたIVDだけではカバーしきれないという点では日本も米国も同じです。しかし、米国はCLIAという制度があるので、LDTに対してある程度の質保証が可能ですが、日本は精度の低いhome-brew assayを排除する制度がないところが問題ですね。
登:精度管理が可能な仕組みが必要ですが、日本ではCLIAのような法律をつくること自体が非常に難しいと思います。
|
||
| ▲ このページのトップへ |