����������Â̍L�� GIcancer-net



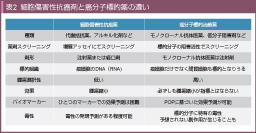
�@�����q�W�I��́A���זE�̑��B�E�i�W�Ɋւ��d�v�ȕ��q (�Q) �ڕW�I�Ƃ������Ö�ł���A�]���̍זE���Q���R���܂ƑΔ䂵���ď̂ł���B���̈Ⴂ��������������B�זE���Q���R���܂̖�܃X�N���[�j���O�́A��ʓI�Ɋ��זE����p�������B�A�b�Z�C�ɂčs�����A�����q�W�I��ł͕W�I���q�̑j�Q�����ɂ��X�N���[�j���O����邱�Ƃ������B�܂��A�זE���Q���R���܂̕W�I�͊��זE�̊j�_ (DNA�ARNA) �ł��邪�A�����q�W�I��̕W�I�͗l�X�ł���A���זE�����łȂ����g�D���ӂ̌��Ǔ���זE�Ȃǂ̊Ԏ��זE���W�I�ƂȂ蓾��B����̓_�ł��A�זE���Q���R���܂͊��̏k�������ʂ̎w�W�ƂȂ邪�A�����q�W�I��ł͕K��������ᇏk���ƌ��ʂ����ւ���Ƃ͌��炸�A�����̑��B�}�����ʂ݂̂�L�����܂������B�܂��A�����q�W�I��͊����ٓI�ȕ��q��W�I�Ƃ��邽�߁A��ᇑI�𐫂������Ő����y�x�ł��邱�Ƃ����҂����B�������A�����q�W�I��ł́AEGFR�j�Q�܂ɂ����]�A�Ԏ����x����VEGF�j�Q�܂ɂ�鍂�����A�n�������x���A�`���A�A�����ǐ��ǁA�����ǐ��E�ȂǁA�W�I�ƂȂ镪�q�ɓ��L�̓Ő���J���i�K�ł͗\�z����Ȃ��Ő���������B
�@�����q�W�I��ł́A�W�I���q�����炩�Ȃ��߂ɉȊw�I�����Ƃ��Ă�POP (proof of principle) �����߂��A���ʂ����҂ł���W�c�𑁊�������肵�A���̃o�C�I�}�[�J�[��p���ėՏ��������s�����Ƃ����҂����B�������A�����q�W�I��̗��j��U��Ԃ�ƁA�����͊J����������̃o�C�I�}�[�J�[�������Ȃ�������ɂ��ꂽ���߁A�����I�ȊJ�����s���Ȃ������B���̔��Ȃ���A2005�N�ɕč�FDA�ɂ����Drug-Diagnostic Co-Development�\�z������A���݂ł͌��ʗ\���f�f�@ (�R���p�j�I���f�f��)�Ɩ�܊J���Ƃ������i�s�ōs����J���헪����������Ă���B
�@�ᕪ�q�����q�W�I�R���܂͊��זE���̃V�O�i���`�B���q��W�I�Ƃ��đj�Q���ʂ������A������ʎY����r�I�e�Ղł��邱�Ƃ���o�������B2001�NImatinib���ŏ��Ɉ��i�Ƃ���FDA���F����A���ݎg�p����Ă���ᕪ�q�����q�W�I�R���܂̑������A�L�i�[�[��W�I�Ƃ���L�i�[�[�j�Q�܂ł���B�L�i�[�[��ATP�̃����_����A�~�m�_�c��ɂ���q�h���L�V��Ɉړ������A���L���������銈����L����B�܂��A�L�i�[�[���g�̊��������߂��������g�̃����_���ɂ���čs���� (���ȃ����_��)�BGefitinib�AImatinib�Ȃǒᕪ�q�L�i�[�[�j�Q�܂̂قƂ�ǂ�ATP�������ʂɋ����I�ɑj�Q���邱�ƂŃV�O�i���`�B���Ւf������̂������B����ŁAMEK�j�Q�܂ł���Trametinib�̓L�i�[�[�̊������ʂƂ͕ʂ̏ꏊ�Ɍ������邱�Ƃōy�f������ቺ������ (�A���X�e���b�N����) �������ł���B
�@�ᕪ�q�����q�W�I�R���܂̊J���ł́A�܂����q�����w�I�ȗ��_�Ɋ�Â��ĕW�I���q�����肵�A���̕��q�ɑ_�����߂Ċ�����j�Q���鉻�������e�����Ђ������������C�u�����[�̒�����T���o����Ƃ��s���� (�n�C�X���[�v�b�g�X�N���[�j���O)�B���邢�́A�W�I���q�̗��̓I�\���╨�����w�I��������A�ǂ��悤�ȉ��������������ʂɓK�����邩���l�����Đv���Ă������@������B�����̎�@�ɂ�蓾��ꂽ��������L�͌�� (���[�h������) �Ƃ��āA����ɗL�����A�I�𐫁A���ԂȂǂ����ǂ��Ă����Ȃ���œK�����Ă����B�ᕪ�q�����q�W�I�R���܂̌��_�Ƃ��āA���Ȃ��炸�W�I���q�ȊO�ɂ���p����\�������� (�I�t�^�[�Q�b�g����)�A���ꂪ�\���ł��Ȃ��Ő��Ɍq���邱�Ƃ����邽�ߓ��ɒ��ӂ�v����B
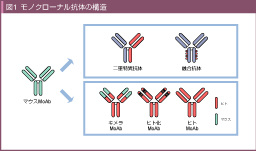
�@�R�̂Ƃ́A����̕��q (�R��) ��F�����Č�������y���Əd������Ȃ�l�ʑ̃^���p�N�ł���B�R�̈���������@���́A�@�R���ł���W�I���q�Ɍ������邱�Ƃɂ���ĕW�I���q�̋@�\�𐧌䂷��A�A�R�̂��L����R�̈ˑ����זE���Q���� (antibody-dependent cellular cytotoxicity: ADCC) ���̈ˑ����זE���Q���� (complement-dependent cytotoxicity: CDC) �Ȃǂ̃G�t�F�N�^�[�����ɂ���ĕW�I���q������זE����������A��2�ɑ�ʂ����B�R�̈��̃A�C�\�^�C�v�́A�G�t�F�N�^�[������L���Ĕ��������������Ƃ���IgG���L���p�����A�T�u�N���X�̓G�t�F�N�^�[�������K�v�ȏꍇ�ɂ�IgG1���A�t�ɕs�v�ȏꍇ�ɂ�IgG2��IgG4���p������B
�@1970�N���Lindemann��Jerne�炪����idiotypic network theory�Ɋ�Â�1, 2)�A1980�N���CEA��17-A1 (EpCAM) �Ȃǎ�ᇊ֘A�R���ɑ���}�E�X���m�N���[�i���R�̂ɂ��R�̗Ö@�����݂�ꂽ�B17-A1�ɑ���}�E�XIgG2a�R�̂�Edrecolomab�̗Տ��������s���A���̓��ł̍Ridiotypic�R�̗̂U�����m�F���ꂽ���A�������p��⏕���w�Ö@�Ƃ���5-FU/LV�Ö@�Ƃ̕��p������������III�������ł͏�悹���ʂ������Ȃ�����3)�B
�@�}�E�X���m�N���[�i���R�̂������q�W�I��Ƃ��ėp����ꍇ�A�q�g�ɑ���R������L���邱�Ƃ���HAMA (human anti-mouse antibody) ���U������A�R�̂̊�������┼�����Z�k�������_�A�A�i�t�B���L�V�[���p�^������ȓ_�����ƂȂ����B�����ŁA�}�E�X�R�̂̒��̈���q�g�R���̂��̂ɒu���������L�����R�̂��J�����ꂽ�B�L�����R�̂́A�ϗ̈悪�}�E�X�R���̂��ߍR���������͕ێ�����AHAMA�̏o���p�x���Ⴍ�A�����������������Ƃ��������_��L���Ă���A1997�N��Rituximab�����������p��ɑ���R�̈��i�Ƃ��ď��߂ď��F����Ă���B����Ńq�g�R�L�����R�� (human anti-chimeric antibody: HACA) �̏o����d�Ă�infusion reaction�����ꂽ���߁A���������̖Ɖu�����ቺ��ړI�ɁA�R���������ʂł��鑊�����̈� (complementarity determining region: CDR) �ȊO���q�g�^�ɒu�������q�g���R�̂��J�����ꂽ�B���̌�1990�N��㔼�ɁA���S�Ƀq�g�R�̂̍\����L���銮�S�q�g���R�̂̍쐬���\�ƂȂ��Ĉȍ~�A�R�̈��̎嗬�ƂȂ��Ă���B
�@����ɁA�R�̍H�w�̔��W�ɂ��l�X�ȍH�v���{���ꂽ�R�̈�o�ꂵ�Ă���B2009�N�ɉ��B�Ŋ��������̎��Ö�Ƃ��ď��F���ꂽCatumaxomab�́A�����p���\�ʕ��qCD3�Ǝ�ᇍR��EpCAM�̑o����W�I�ɂ����d���ٍR�̂ł���A�Ǐ��ɃG�t�F�N�^�[�זE���W�ς��邱�Ƃő�����ʂ����҂ł���B�܂��A�R�̂ɍR���܂���ː����ʌ��f�Ȃǂ�����������ᇂ�I��I�ɏ��Q������Z���R�̂̊J��������ł���BT-DM1��HER2�̍R�̈��ł���Trastuzumab�ɍזE���Q���R����DM1�����������\�������Ă���A�זE���ɋz��������DM1�����o����ĎE�זE���ʂ���������A�����ł�Trastuzumab��������ʂ�����Ă���4)�B�܂��A���̈�̃A�~�m�_�z���ω��������蓜���C�����{�����肷�邱�ƂŃG�t�F�N�^�[���ʂ����߂Ă���R�̈�������B�Ⴆ�AMogamulizumab�͐��lT�זE�����a�����p�� (ATL) �זE�̕\�ʍR���ł���CCR4�ɑ���R�̈��ł��邪�AADCC���������߂�ړI�ōR�̂��ۗL���铜���̒��̃t�R�[�X��ቺ�������R�̂ɂȂ��Ă���B
�@���ݏ��F����Ă�������q�W�I��̑������ᕪ�q�����q�W�I�R���܂����m�N���[�i���R�̈��ł��邪�A�z�������Ö@�ŗp�����������Ö@�܂�A�}���O�������������a (APL) �ɗp������S�g�����X�^���`�m�C���_ (ATRA)�A������������ŗp������T���h�}�C�h�n��܂������q�W�I��Ɋ܂܂��ꍇ������B�܂��A��`�q�H�w�Z�p�̐i���ɂ��R�̈��ȊO�̃o�C�I���i���o�ꂵ�Ă���B�Ⴆ��Aflibercept��VEGFR-1��VEGFR-2�̍זE�O�h���C���̈ꕔ���q�gIgG1��Fc�̈�ɗZ����������e��/IgG�R��Fc�Z���^���p�N�����i�ł���A��������Ώۂɑ�III���������s���Ă���Trebananib (AMG386) �́A�A���W�I�|�G�`��-1, 2�Ɍ�������y�v�`�h�ƃq�gIgG1��Fc������Z���������^���p�N�����i (�y�v�`�{�f�B) �ł���B�Ȃ��A���זE�̕\�ʍR�� (��ᇍR��) ��Ɖu�S���זE�ɔF�������čR��ᇌ��ʂ����y�v�`�h���N�`���́A�����q�W�I��ƌĂ��ꍇ�����邪�A�Ɖu�S���זE������ԐړI�ȕW�I���Âł��邽�߁A(���`��) �����q�W�I��ɂ͊܂܂�Ȃ��B
�@�����q�W�I��̕W�I�ƂȂ镪�q�����邽�߂ɂ́A���̓����A�܂���Ɛ���g�D�Ƃ��ǂ̂悤�ɈقȂ�̂��𖾂炩�ɂ��邱�Ƃ���n�܂�BHanahan��Weinberg��2000�N��Cell���ɁuHallmarks of Cancer�v�Ƒ肵�ă��r���[���A���̃z�[���}�[�N (����) �Ƃ��āASelf-sufficiency in growth signals, Insensitivity to anti-growth signals, Tissue invasion & metastasis, Limitless replicative, Sustained angiogenesis, Evading apoptosis��6��������5)�B�����āA���ꂼ��̓������i���Ă��镪�q���A�����Â̕W�I���q���ƍl�����A����ɕ��q�W�I��̊J�����s��ꂽ�B�Ȃ��A2011�N�ɂ́uHallmarks of Cancer: The Next Generation�v�Ƒ肵�ăA�b�v�f�[�g����Ă���A���̒��ł́A2000�N�ȍ~�̊������̐��ʂ܂��A�V����4�̃z�[���}�[�N���lj�����ASustaining proliferative signaling, Evading growth suppressors, Avoiding immune destruction, Enabling replicative immortality, Tumor-promoting inflammation, Activating invasion & metastasis, Inducing angiogenesis, Genome instability & mutation, Resisting cell death, Deregulating cellular energetics��10���ڂ��������Ă���6)�B
�@���ݏ��F����Ă����܂̔����ȏオ�L�i�[�[��W�I�Ƃ��Ă���B�L�i�[�[�̓A�~�m�_�̂�����ɃZ�����A�X���I�j���A�`���V���c��������_�������邪�A���̂�����90%���Z�����A��10%���X���I�j���ł���A�`���V����0.05%���x�ɉ߂��Ȃ��B�������`���V���̃����_���������w�I�ɏd�v�ȃP�[�X�������A�����Â̕W�I�ƂȂ邱�Ƃ������B�W�I�ƂȂ�`���V���L�i�[�[�Ƃ��ẮAEGFR�AHER2�AVEGFR�AIGF1R�AFGFR�Ac-Kit�Ac-Met�Ȃǂ̎�e�̌^�`���V���L�i�[�[��ASrc�AJAK�ABcr-Abl�Ȃǂ̔��e�̌^�`���V���L�i�[�[������A������W�I�Ƃ�����܂��J���E�Տ����p����Ă���B�܂��A�Z����/�X���I�j���L�i�|�[�Ƃ��Ă�PI3K�AAkt�AmTOR�ACDK�APLK�AMEK�ARaf�Ȃǂ�W�I�Ƃ������ÖJ���E�Տ����p����Ă���B�L�i�[�[�ȊO�̕W�I�Ƃ��ẮAHDAC�j�Q�܁A�E���`�����܂Ȃǂ̃G�s�W�F�l�e�B�N�X�֘A��e�������[�[�j�Q�܁A�A�|�g�[�V�X�U���܁AHedgehog�j�Q�܁ANotch�j�Q�܂Ȃǂ���������B���̂悤�Ɋ��̕W�I���q�͔��ɑ���ɂ킽��B���זE�����łȂ����g�D���`�����Ă��錌�Ǔ���זE�A���ǍזE�A���ۉ�זE�Ȃǂ����Â̕W�I�ƂȂ蓾�邽�߁A������A�����̐i���ɔ����V���ȕW�I���q�̌�₪�����Ă����ƍl������B
GI cancer-net
����������Â̍L��