消化器癌治療の広場 GIcancer-net



大腸癌では約40%にKRAS変異を認め、Rasの恒常的活性化が認められる。よって、Rasを標的とした分子標的治療が期待されたが、Rasは低分子結合タンパクの一種で、不活化状態ではGDPと結合しており、主にファルネシルトランスフェラーゼ (FT) によってファルネシル化され活性化される。このFTの阻害剤がRasの働きを阻害する分子標的薬として臨床開発が行われた。
TipifarnibはFTの選択的阻害剤である。難治性固形癌を対象とした用量漸増第I相試験が行われ (28例) 87)、400mg bidにて4例中2例にDLT (grade 4の好中球減少) が生じ、RDは300mg bidとされた。主な有害事象は、悪心・嘔吐、疲労、骨髄抑制であった。なお、大腸癌の1例において50%以上のCEA低下と若干の腫瘍縮小を認めたことから、難治性大腸癌を対象に第III相試験が行われ、368例の患者がTipifarnib群とplacebo群とに2:1で割り付けられた88)。主要評価項目のOSは中央値Tipifarnib 群174日、placebo群185日と両群に差を認めず (p=0.376)、PFS中央値もTipifarnib 群81日、placebo群80日と差を認めなかった。また、KRAS codon 12変異の有無別の解析も行われたが、KRAS変異の有無と効果との間に交互作用は認めなかった。以上から、大腸癌に対するTipifarnibはKRAS変異の有無にかかわらず効果が乏しいと考えられた。基礎実験ではゲラニルゲラニルトランスフェラーゼなど他の酵素がRasの活性化を代償することも示されており、FT阻害だけではRasの活性化が阻害されないのかもしれない。
SorafenibはVEGFR、PDGFR、c-KIT、RETなどのチロシンキナーゼやBRAF、Raf1 (CRAF) などのセリンスレオニンキナーゼに対する阻害作用を有する、分子量464.825の低分子性分子標的抗癌剤である。1994年にバイエル社とオニックス社がRaf1の活性阻害を指標として行った化合物スクリーニングの際に発見された。その後、基礎研究にてRaf1以外にもVEGFR-1、VEGFR-2、VEGFR-3、PDGFR-1、c-KIT、RET、BRAF、BRAF V600Eなどを低濃度で阻害することが明らかとなった89)。69例の難治性固形癌患者を対象にした第I相試験においてSorafenib (50〜800mg 1日1〜2回投与) の安全性と薬理学的動態が検討された結果、最も頻度の高い有害事象は下痢 (55%) であり、DLTは800mg bid群でgrade 3の下痢と疲労、600mg bidでgrade 3の皮膚毒性を認めた90)。その結果、MTDは400mg bidであった。薬物動態解析では薬物動態の個人差が大きく、400mg bid以上での有害事象頻度は用量依存的ではなかった。薬力学的解析では、200mg bid以上で末梢血リンパ球のERK活性阻害効果が認められた。抗腫瘍効果の探索的検討では、肝細胞癌1例でPR、腎細胞癌1例で約1年以上のSDが得られたことから、両疾患を優先して臨床開発が行われることとなった。なお、本試験では大腸癌患者が26例登録されており、PR例は認めなかったものの6ヵ月以上SDが4例、うち2例が1年以上のSDが得られた。
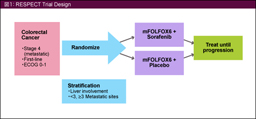
SorafenibはVEGFRなど血管新生に関連した因子だけでなく、BRAF、RAF1などEGFR-RAS下流に位置する増殖因子の阻害作用も有することから、大腸癌、特にKRAS変異型の大腸癌への効果が期待された。大腸癌初回治療におけるmFOLFOX6 + placebo療法とmFOLFOX6 + Sorafenib療法とを比較した第II相試験 (RESPECT試験) では、主要評価項目であるPFSの中央値はplacebo群 (101例) 8.7ヵ月、Sorafenib群 (97例) 9.1ヵ月であり、有意差を認めなかった (HR=0.884, 95% CI: 0.635-1.231, p=0.2309) 91)。KRAS変異型に対してもplacebo群7.6ヵ月、Sorafenib群8.9ヵ月と、Sorafenib群でやや良好であったものの有意差は認めなかった (HR=0.895, p=0.34)。Grade 3以上の有害事象として好中球減少、口内炎、下痢、PRE (可逆性後白質脳症) 症候群などがSorafenib群で多くみられ、治療中止理由は病勢進行例がSorafenib群で少なく (placebo群64%、Sorafenib群51%)、有害事象 (6% vs. 9%) や患者希望 (12% vs. 22%) による中止が多かった。
また、CPT-11やCetuximabとSorafenibとの併用療法も検討が行われている。Mrossらは固形癌を対象としたCPT-11 + Sorafenib (400mg/day) 療法の第I相試験を行い、CPT-11未使用例14例に対して77%でSDが得られ、有害事象も忍容可能であったと報告している92)。SamalinらはKRAS変異型の大腸癌に対する2nd-line以降としてCPT-11 + Sorafenib療法の第I/II相試験 (NEXIRI試験) の結果を報告している93)。第I相部分には10例が登録されたがDLTは認められず、CPT-11 (180mg/m2 隔週投与) とSorafenib (400mg bid) により第II相部分が行われた。CPT-11投与歴のあるKRAS codon 12または13変異を有する54例に治療が行われ、主要評価項目の病勢コントロール率は64.9% (PR1例を含む) であった。また、PFS中央値は3.5ヵ月、OS中央値は7.7ヵ月であり、KRAS変異型でCetuximabやPanitumumabを検討した他の試験よりも良好であることから、効果が期待されると考えられた。現在、CPT-11を含む治療に不応となった患者を対象にCPT-11 + Sorafenib療法をCPT-11単独療法、Sorafenib単独療法とを比較する無作為化第II/III相試験 (NEXIRI2試験) が行われている94)。なお、NEXIRI試験ではgrade 3/4の有害事象として下痢39%、手足症候群15%、好中球減少35%と毒性が強かったことから、NEXIRI2試験でのCPT-11 + Sorafenib療法は1コース目のCPT-11投与量を120mg/m2に設定し、grade 2以上の毒性がなければ2コース目以降に150、180mg/m2と増量していくレジメンとなっている。
また、Bevacizumabとの併用も検討され、標準治療に不応となった大腸癌を対象にSorafenib (200mg bid 5日内服2日休薬) とBevacizumab (5mg/kg 隔週投与) の併用療法の第II相試験では、評価可能であった79例で奏効率2.5%、病勢コントロール率63%、PFS中央値3.1ヵ月、OS中央値 6.7ヵ月との結果が報告されている95)。
Sorafenibによる治療開発は継続されているが、後述するように同じバイエル社から同系統の薬剤であるRegorafenibが大腸癌に対する承認を取得したことから、大腸癌領域におけるSorafenibの位置づけは難しいものとなっている。
Regorafenibは、Sorafenibと同じくビアリール尿素誘導体の低分子性分子標的抗癌剤である。腫瘍増殖に関連するBRAF、Raf1などのセリンスレオニンキナーゼに加え、腫瘍血管新生に関わるVEGFR-2、VEGFR-3、TIE-2、PDGFR、FGFR1などのチロシンキナーゼ、p38αMAPキナーゼ等を標的とするマルチキナーゼ阻害剤である。大腸癌、乳癌、腎癌細胞株の移植腫瘍モデルでは、10mg/kgと低用量Regorafenibの1日1回経口投与で腫瘍血管密度低下と腫瘍抑制効果が認められた96)。標準治療に不応となった固形癌患者を対象とした第I相試験が行われ、53例に10〜220mgのRegorafenib (21日内服7日休薬) が投与された97)。220mg群で12例中5例に1サイクル (28日) 以内でのDLT (手足症候群、腹痛など) を認め、2サイクル以内に減量が必要であった例は12例中9例であった。一方、160mg群では2サイクル以内での減量は12例中3例であった。以上から160mg/dayがRDと決定された。頻度の高い有害事象は皮膚毒性 (手足症候群、皮疹を含む)、高血圧、下痢であり、血中のRegorafenibと代謝活性産物の薬物血中濃度―時間曲線下面積 (area under the blood concentration time curve: AUC) は概ね用量依存的であった。なお、評価可能であった47例のうち大腸癌1例を含む3例で奏効が認められた。さらに、標準治療不応後の大腸癌患者を対象とした第I相試験では、60〜220mgのRegorafenib (21日内服7日休薬) が38例に投与された98)。評価可能であった27例について抗腫瘍効果の探索的な検討を行ったところ、PRが1例、SDが19例に得られPFS中央値は107日であった。
これらの結果をもとに、標準治療不応の大腸癌患者を対象にRegorafenibの有効性を検証するプラセボ対照第III相国際共同治験 (CORRECT試験) が行われた99)。結果、主要評価項目であるOSの中央値はplacebo群5.0ヵ月、Regorafenib群6.4ヵ月とRegorafenib群で有意な延長を認め (HR=0.77, p=0.0052)、PFS、病勢コントロール率も同様にRegorafenib群で良好であった。また、KRAS status別の解析では、KRAS野生型 (299例) でのOS中央値はそれぞれ5.0ヵ月、7.3ヵ月 (HR=0.653)、KRAS変異型 (430例) ではそれぞれ5.1ヵ月、6.2ヵ月 (HR=0.867) であった。主な有害事象は、手足症候群、疲労、下痢、高血圧、皮疹などであった。この結果より、FDAは標準治療後の大腸癌に対する治療においてRegorafenibを2012年9月に承認した。CORRECT試験には本邦からも100例が登録されており、欧米と比較して毒性発現頻度の違いが示唆されたものの忍容可能で、効果に大きな違いは認められなかった。本邦でも優先審査の対象に指定され、2013年3月に承認された。
RegorafenibはSorafenibと構造式ならびに標的分子が類似しており、結果として毒性プロファイルも類似している。Regorafenibは単剤療法の第I相試験で奏効例が認められたことから単剤療法での第III相試験が行われ、結果としてplaceboと比較して生存期間の延長が認められた。Sorafenibや他のVEGFRチロシンキナーゼ阻害剤の多くは腎癌、肝癌での開発が先行し、大腸癌では単剤での奏効例が認められず、他の薬剤との併用療法が試みられてきたが、毒性が問題となった。薬剤の違いもさることながら、開発戦略の違いが現在までの試験結果の違いに繋がっている可能性はある。なお、海外では現在、FOLFIRIやmFOLFOX6とRegorafenibの併用療法の臨床試験も行われている100, 101)。
Perifosineは合成アルキルリン脂質の一種であり、1990年代から抗腫瘍効果があることが知られていた102)。抗腫瘍効果の機序は徐々に明らかとなり、現在ではAktの細胞膜への移行を阻害することでAktの活性化を阻害する作用、NF-κBの核内移行を阻害する作用などにより抗腫瘍効果を発揮すると考えられている。固形癌を対象とした3週内服1週休薬のPerifosine単剤療法の第I相試験では、50mg/dayから開始され350mg/dayまで増量したが、DLTは認められなかった103)。しかし、grade 1/2の悪心、嘔吐、下痢の頻度は用量依存性であり早期治療中止の割合も考慮され、200mg/dayがMTDと決定された。また、固形癌を対象にPerifosineの連日投与法 (50〜100mg) と週1回投与法 (900〜1200mg) とを比較した無作為化第II相試験では、連日投与にてgrade 3/4の悪心、嘔吐、下痢の毒性を認めず、連日投与法が妥当と結論された。
2005年からは、Perifosineと様々な抗癌剤との併用療法が試みられた。前治療歴のある大腸、乳腺、前立腺、頭頸部などの固形癌を対象にPerifosineと細胞傷害性抗癌剤との併用療法 (8レジメン) を行うプラセボ対照無作為化第II相試験が行われた。試験は資金の問題で中止されたが、大腸癌25例の解析で、Capecitabine + placebo (CAP) 群と比較してCapecitabine (825mg/m2 2週内服1週休薬) + Perifosine (50mg/day) (P-CAP) 療法の治療成績が良好であったことから、さらに13例が追加され最終解析が行われた104)。結果、TTP中央値はCAP群10.1週、P-CAP群27.5週 (HR=0.254, 95% CI: 0.117-0.555, p<0.001)、OS中央値はそれぞれ7.6ヵ月、17.7ヵ月 (HR=0.370, 95% CI: 0.180-0.763, p=0.0052) と、いずれもPerifosine併用により有意な延長を認めた。この結果を受け、標準治療に不応となったCapecitabineの前治療歴のない大腸癌を対象にP-CAPとCAPを比較する第III相試験 (X-PECT試験) が行われた105)。結果、OS中央値はCAP群6.9ヵ月、P-CAP群6.4ヵ月と、有意差を認めず (HR=1.111, p=0.315)、奏効率、PFSも有意差を認めなかった。有害事象に関しては、grade 1/2の悪心、嘔吐、下痢、疲労の頻度がP-CAP群で高い傾向にあるものの、grade 3/4の有害事象には大きな差を認めなかった。L-OHPを毒性中止したKRAS野生型の集団に限定したサブグループ解析では、PFS中央値がCAP群6.6ヵ月、P-CAP群18.1ヵ月と、P-CAP群で良好であった (HR=0.514, 95% CI: 0.329-0.801, p=0.003)。なお現在、腫瘍組織や血清サンプルを用いたバイオマーカー解析が行われている。
本試験の問題点としては、無作為化第II相試験での結果と第III相試験の結果が大きく異なった点が挙げられ、その要因としてはいくつか考えられている。まず、第II相試験と第III相試験の対象・治療法が異なっている点である。第II相試験では前治療レジメン1つ以上 (2nd-line、3rd-line) の患者が対象であったが、第III相試験では標準治療終了後の患者が対象となっていた。次に、第II相試験のデザインや解析方法 (途中で試験結果をみて症例追加し解析された) が、治療効果の探索的評価として正しく行われなかった可能性が挙げられる。Perifosineの作用機序にはAkt経路阻害以外にも多岐にわたると考えられており、今後の臨床開発のためには治療効果を予測するバイオマーカーの報告が待たれる。
GI cancer-net
消化器癌治療の広場